大家好,作为生信初学者,最近跟着孟叔学习相关知识,有些地方不是很懂,特意前来请教各位大咖!
问题如下:
双端质控结果:
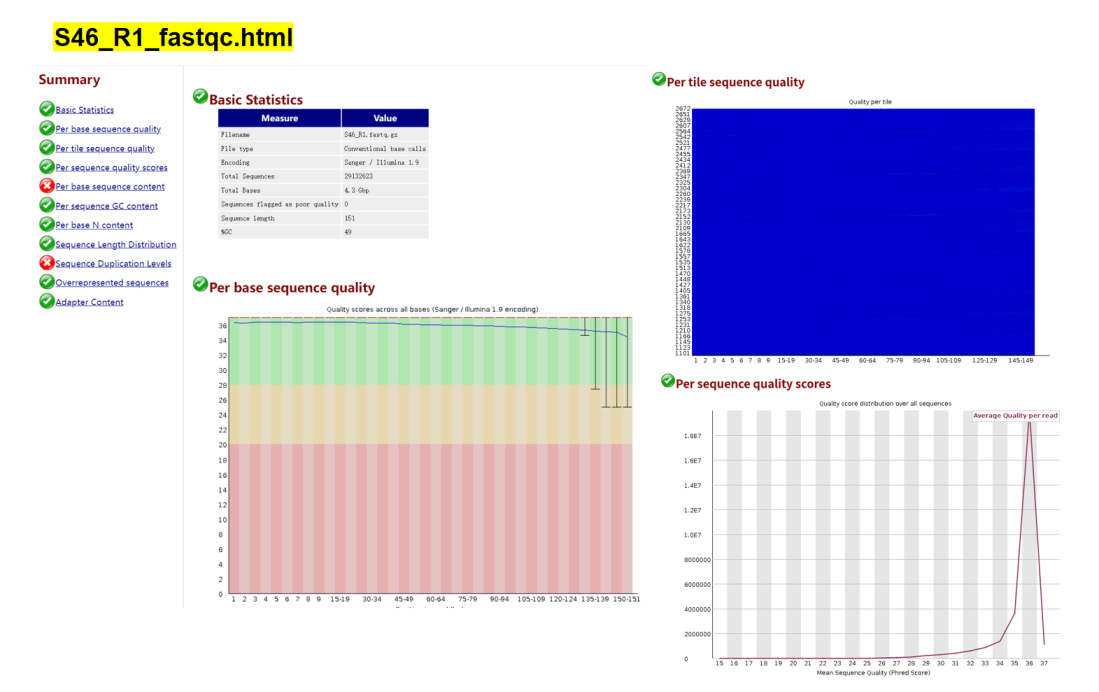
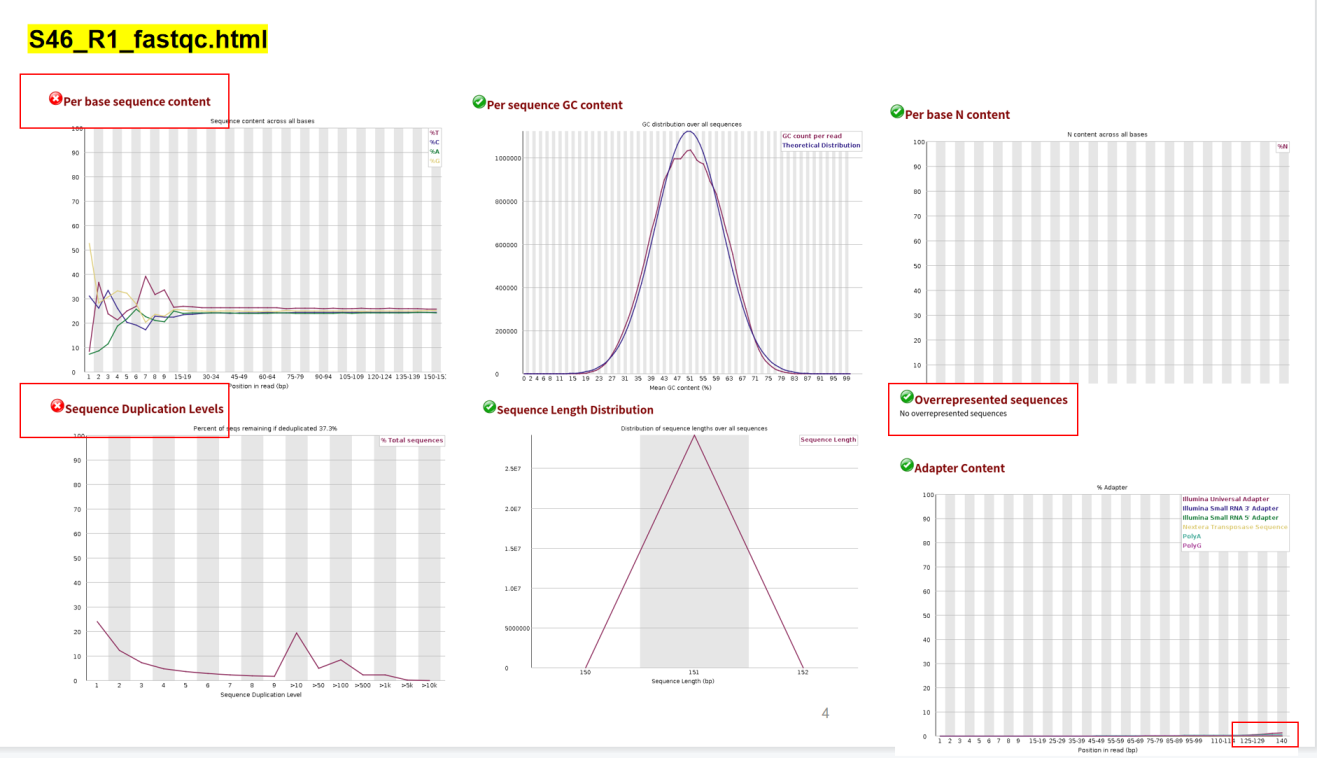
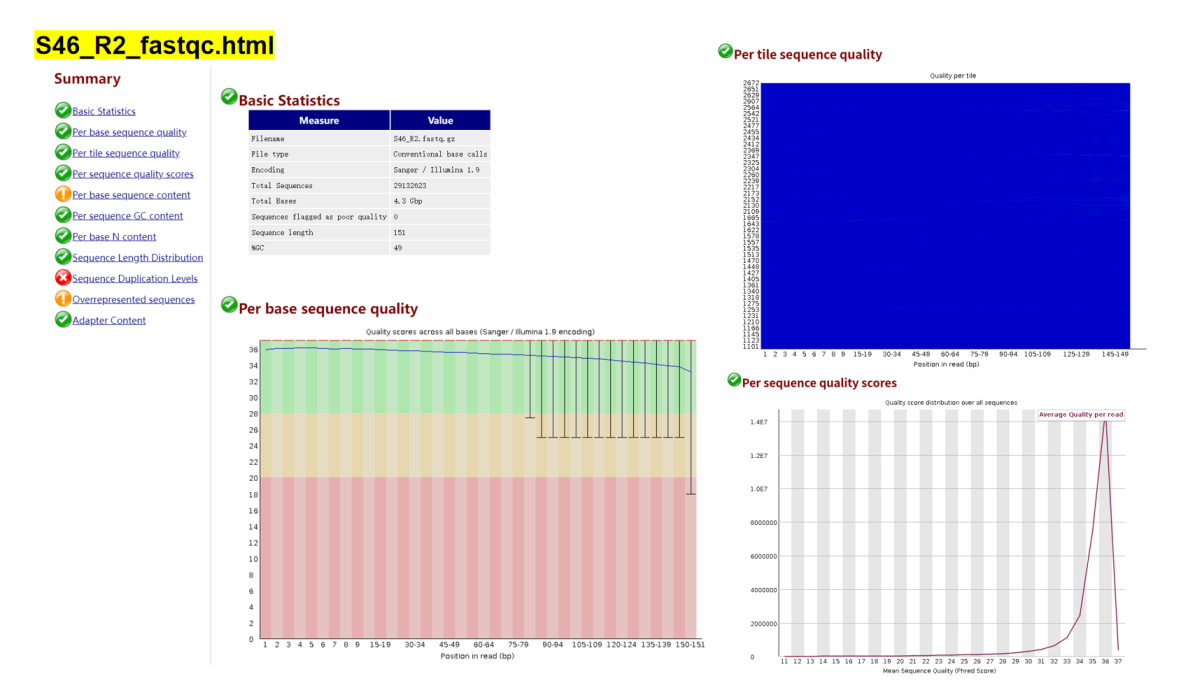
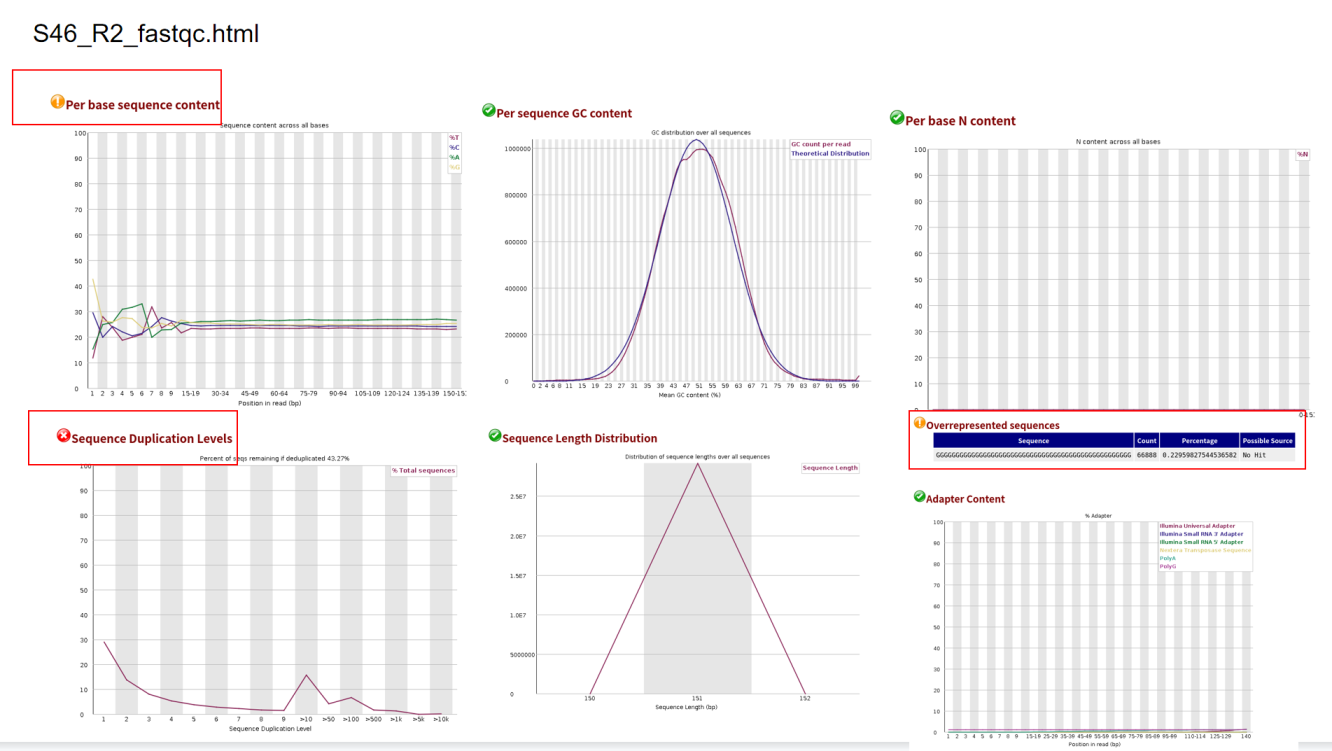
根据以上结果可知,正向测序序列和反向测序序列都有三个不通过指标:Per base sequence content Sequence Duplication Levels Overrepresented sequences 。针对Per base sequence content ,是否有必要切除前面一些10bp序列?在反向测序序列S46_R2_fastqc.html中,Overrepresented sequences提示警告,这是否需要去除?
话句话说,以后在质控结果得知的情况下,一般根据哪几项结果判定是不是可以直接往下做?哪些结果不必要纠结?
用HISAT2进行比对到基因组,自己因为内存原因,没有构建参考基因组索引,直接使用Hisat2官网比对索引文件
这样做是否可以呢,或者说会不会对后面的结果有影响?
做完定量,得到counts表达矩阵,是不是还得做质控?如果是,怎么做质控呀?以及是否要去除线粒体基因?
在此感谢大家的回复!也希望自己的问题能够帮助同样有疑问的人。感谢!
1 回答

这家伙很懒,还没有设置简介