请输入关键字进行搜索
查看更多 "" 的搜索结果
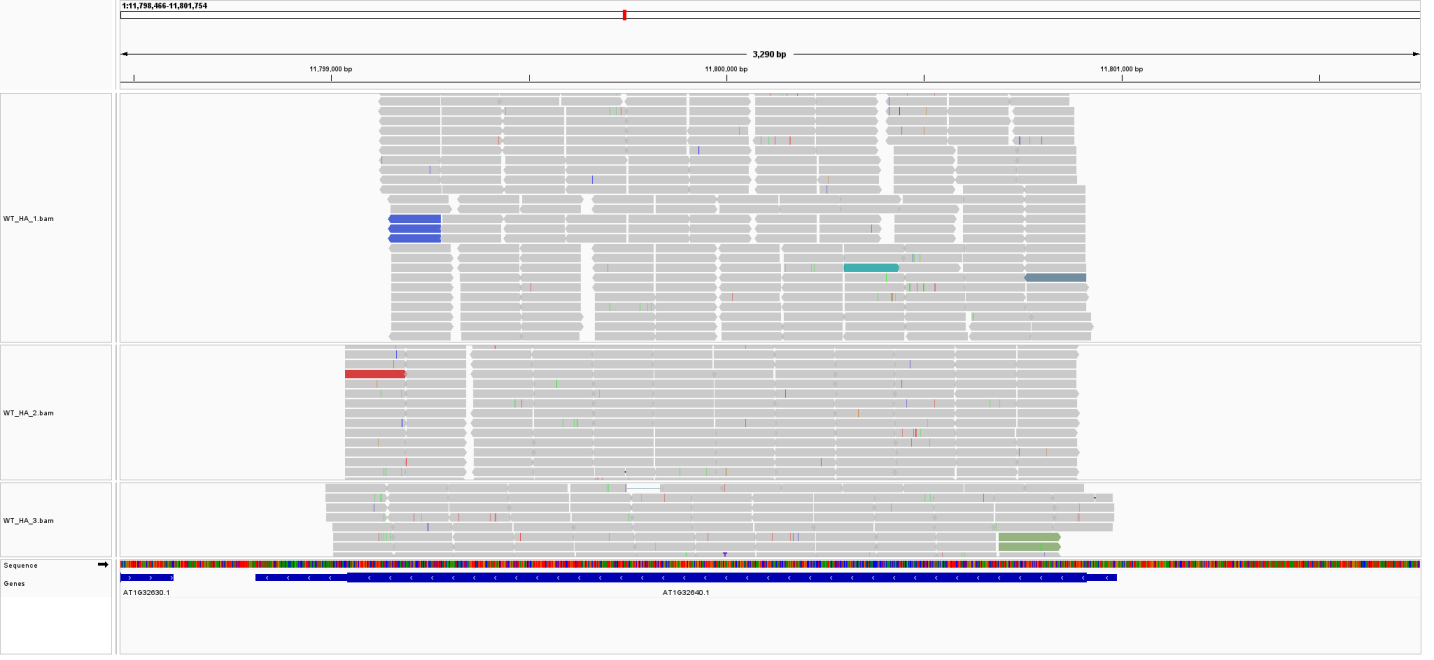
我在服务器上进行转录组分析时(拟南芥),使用hisat2做的比对,用featurecounts进行定量,但是发现有一个基因count和TPM值都为0,但是公司分析结果中这个基因有表达,我使用IGV查看了比对的bam文件,发现是有reads mapping,请问这个原因是?谢谢!
查看全部 3 个回答
这家伙很懒,还没有设置简介
你的浏览器版本过低,可能导致网站部分内容不能正常使用!
为了能正常使用网站功能,请使用以下浏览器