请输入关键字进行搜索
查看更多 "" 的搜索结果
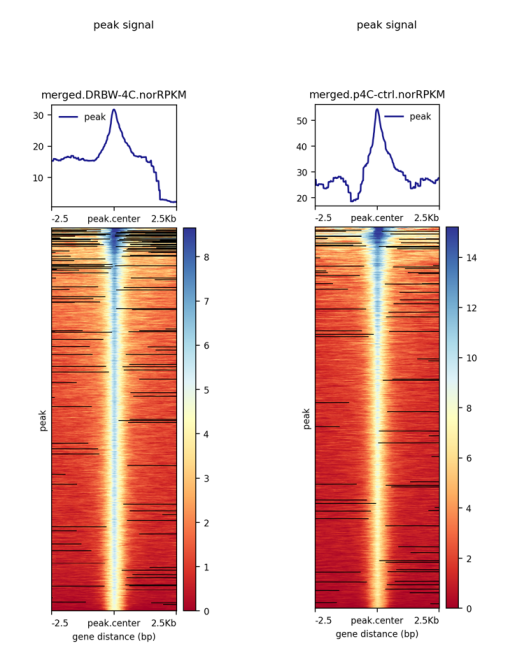
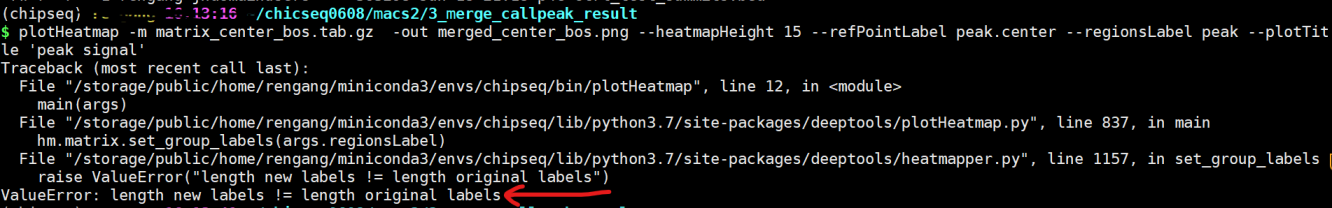
我想把两个样本的曲线合并到一个图里进行比较,之后跑图中所示的两步脚本,在做plotHeatmap这步出现了label的报错信息,求助解决方法,谢谢!
2 回答
这个报错是说你的label长度和你的样品长度不匹配。先去掉这个参数,画一下图即可。最后的名字在Adobe Illustrator里调整。
这家伙很懒,还没有设置简介
你的浏览器版本过低,可能导致网站部分内容不能正常使用!
为了能正常使用网站功能,请使用以下浏览器