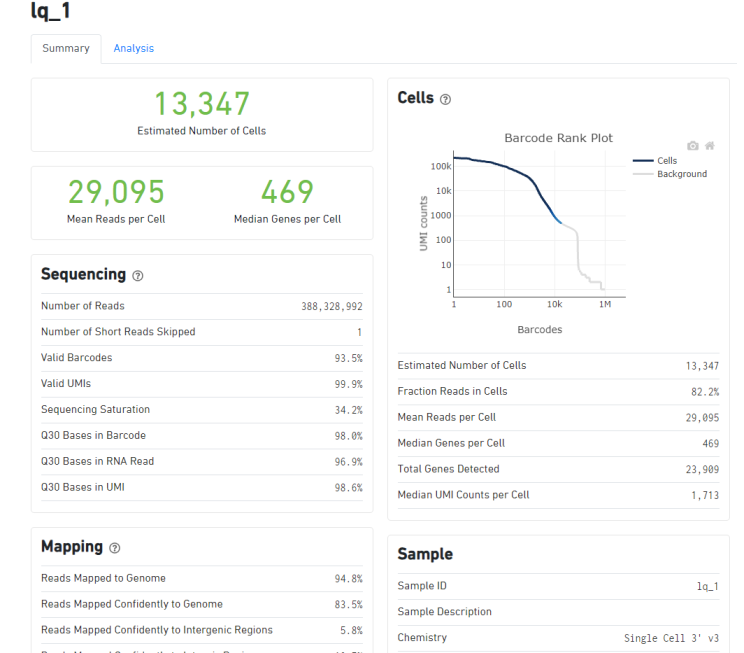
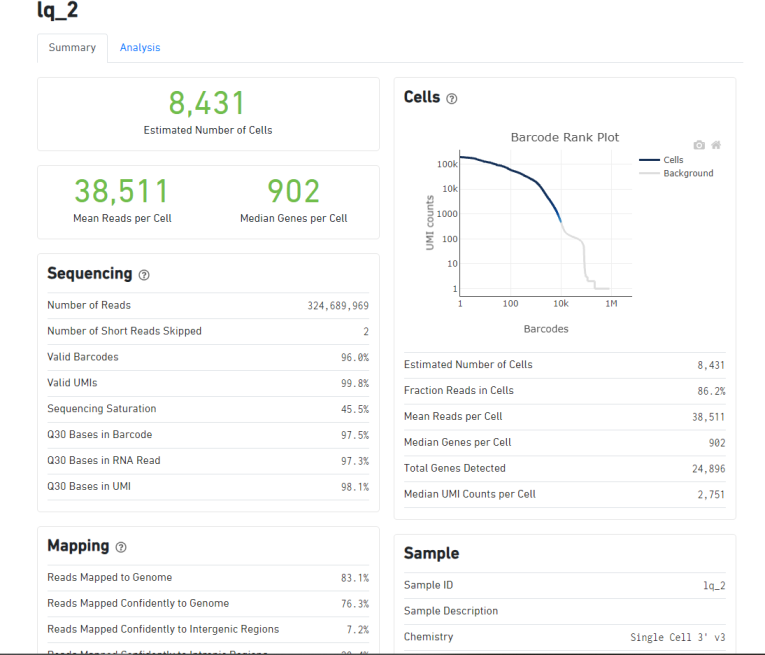
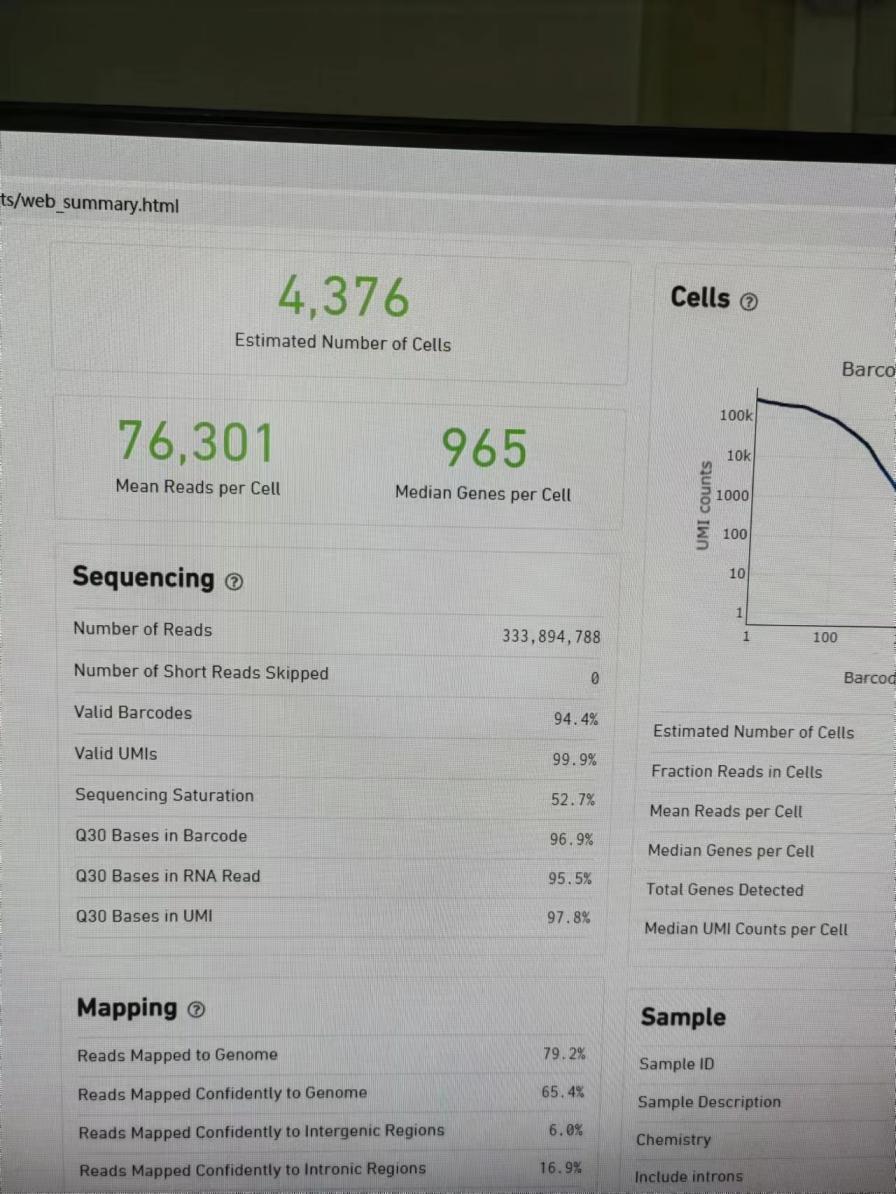
 人源性组织异种移植(PDX)的肿瘤组织的单细胞测序,下机数据用cellranger5.0.1进行分析,代码如下:
人源性组织异种移植(PDX)的肿瘤组织的单细胞测序,下机数据用cellranger5.0.1进行分析,代码如下:
cellranger count --id=lq_2 --transcriptome=refdata-gex-GRCh38-2020-A --fastqs=Fastq/ --sample=lq_2 --localcores=12 --localmem=50,组织是给诺禾进行捕获建库的,当时捕获细胞数是8000个细胞,但是cellranger分析结果如上:
3个样品,其中有两个捕获的细胞数超过了8000个细胞,有一个数据只捕获了4000多个细胞,差异非常大,这样差异大的样品可以一起比较分析吗?这样的数据情况是不是正常的呢?初次做单细胞测序,希望有经验的同学帮忙看一下数据是否有问题,非常感谢。
1 回答
