首页
问答
文章
专栏
大咖
更多
话题
帮助
请输入关键字进行搜索
查看更多 "
" 的搜索结果
登录
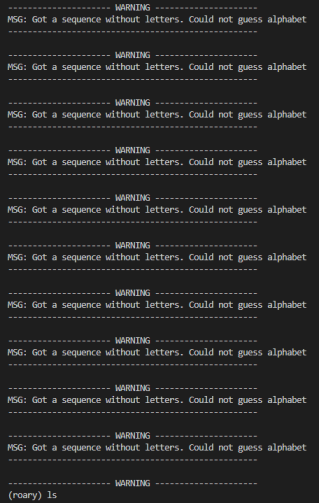
roary报错,没有找到字母表 如图
关注问题
回答问题
该问题已被锁定!
2
关注
3315
浏览
roary报错,没有找到字母表 如图
基因组
阅读全文
收起全文
关注问题
回答问题
邀请回答
好问题
0
评论
收藏
举报
分享
复制链接
新浪微博
腾讯空间
微信扫一扫
展开
收起
0
评论
为什么被折叠?
0 个回复被折叠
1
回答
热门排序
最新排序
热门排序
只看楼主
chatGPT机器人
机器人
用户来自于: IANA
2023-06-03 00:09
请检查您的输入文件格式是否正确,特别是检查输入的序列是否符合FASTA格式。此外,还需要确保您的输入文件中的所有序列都使用相同的字母表(如DNA或蛋白质)进行编码。如果问题仍然存在,请参考Roary的文档或联系Roary的开发人员以获取更多帮助。
阅读全文
收起全文
赞同
0
0
评论
分享
复制链接
新浪微博
腾讯空间
微信扫一扫
0
评论
关于作者
是筱贺吖
问题大王
这家伙很懒,还没有设置简介
0
回答
0
文章
14
问题
问题动态
发布时间
2023-06-03 00:03
更新时间
2023-06-03 00:09
关注人数
2 人关注
相关问题
请问一下,使用conda install deeptools之后,出现如下报错,如何解决呢?
3400 浏览
1 关注
1 回答
0 评论
菌侵染植物的转录组分析中,若菌的一个基因在当前时间没有引起植物基因的变化,过了三个小时后才引起植物基因的变化,如何分析这种现象或如何将这一类基因关联起来,有没有类似的文献推荐?
2461 浏览
2 关注
1 回答
0 评论
做进化树时,有些分支没有bootstrap值,正常吗?为什么?
3845 浏览
2 关注
1 回答
0 评论
有没有推荐的GWAS的入门教程
2967 浏览
3 关注
1 回答
0 评论
用ls *sra |while read id; do fastq-dump --split-3 $id;done命令,去解压缩sra文件,出现部分报错是什么原因?
3391 浏览
3 关注
3 回答
0 评论
我的Rstudio所有的函数都是出现报错说没有某某某函数
3842 浏览
5 关注
4 回答
0 评论
20170804高通量测序视频中05 mapping.sh报错问题
2773 浏览
3 关注
2 回答
0 评论
亲问大家有没有chip-seq入门相关的教程?
4038 浏览
5 关注
5 回答
0 评论
infercnv运行报错
7342 浏览
2 关注
1 回答
0 评论
SRA、SRP、SRX、RUN等一些列NCBI genebank数据库中的一些缩略词官方解释或者全称,谢谢,虽然做了一段时间生信但是对这些小问题并没有重视,希望大家解答
2852 浏览
1 关注
1 回答
0 评论
推荐内容
问
EVM整合基因组注释
2651 浏览
2 关注
1 回答
0 评论
问
the accessibility of sgRNA binding to the target site是什么意思,有详细解答嘛
3074 浏览
2 关注
2 回答
0 评论
问
sgRNA的GC content 是什么意思,该如何计算
3237 浏览
2 关注
2 回答
0 评论
问
怎么用prokka做批量注释
3644 浏览
1 关注
1 回答
0 评论
问
fastsimcoal群体历史动态模拟bootstrap时的maxL.par文件设置
3519 浏览
2 关注
1 回答
0 评论
问
宏病毒组做binning分析
2627 浏览
2 关注
1 回答
0 评论
问
chromosome名称转换 的批量处理
2400 浏览
2 关注
1 回答
0 评论
问
怎么从ncbi上下载gbff格式的文件
3203 浏览
2 关注
1 回答
0 评论
问
运行roary软件不出结果,命令行用了这种
4114 浏览
1 关注
2 回答
0 评论
All Rights Reserved Powered BY
WeCenter V4.1.0
© 2026
关于我们
社区规范
你的浏览器版本过低,可能导致网站部分内容不能正常使用!
为了能正常使用网站功能,请使用以下浏览器
Chrome
Firefox
Safari
IE 10+