如果您在运行Roary软件时没有得到结果,可能有以下几种可能性:
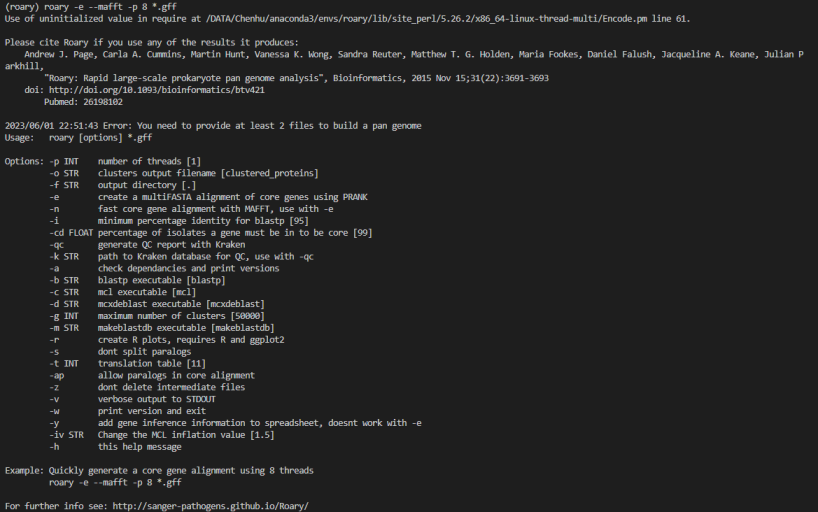
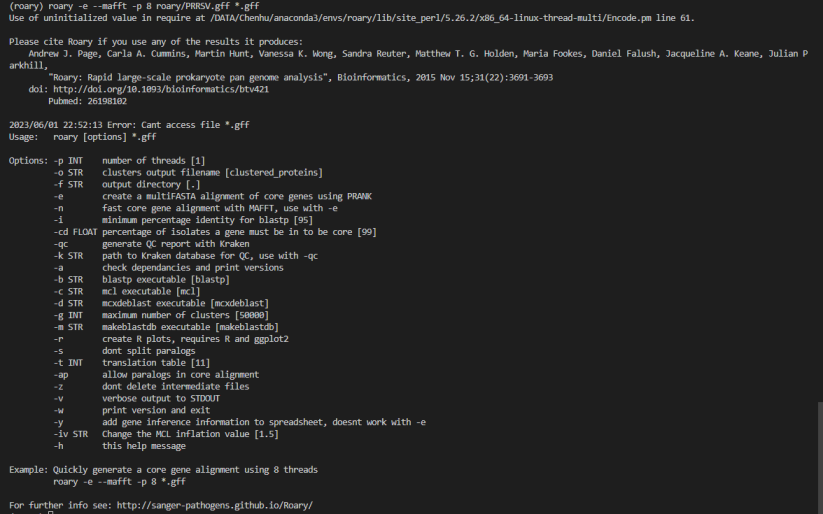
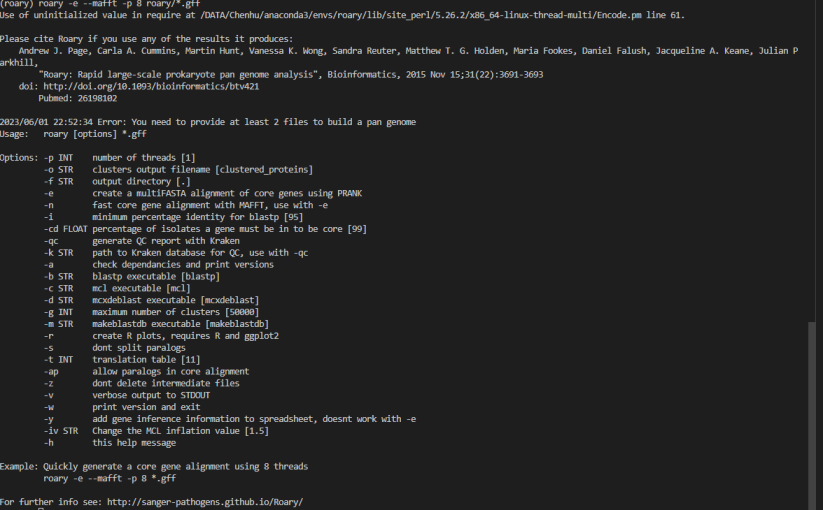
1. 输入文件格式有误:Roary需要输入FASTA格式的基因组序列文件和GFF格式的注释文件。请确保输入文件格式正确无误。
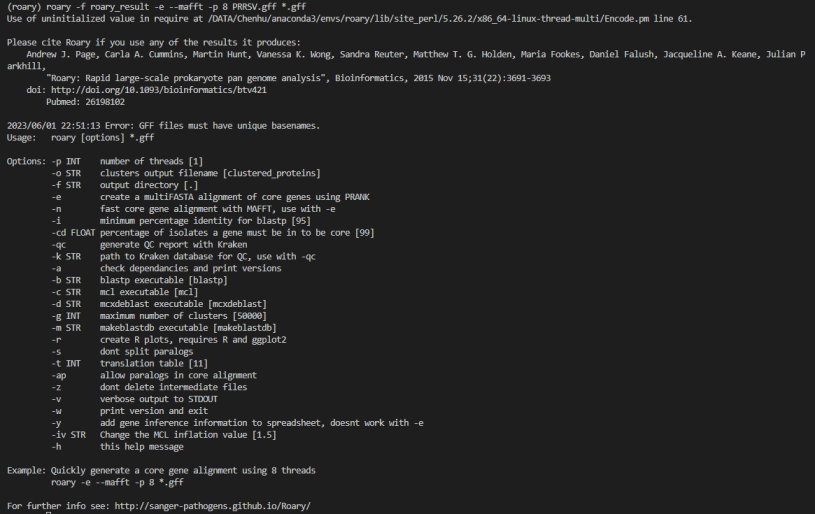
2. 输入文件命名有误:Roary默认按照输入文件名的字典序排序,如果您的文件名不是按照字典序排序的,可能会导致Roary无法正确解析输入文件。请确保输入文件命名正确。
3. 配置文件有误:Roary需要一个yaml格式的配置文件,这个文件包含了Roary的运行参数。请确保配置文件正确无误。
4. 硬件资源不足:Roary需要占用大量的内存和CPU资源。如果您的计算机配置较低,可能无法支持Roary的运行。建议使用高性能计算机或者云计算服务。
以上是可能导致Roary无法正常运行的一些原因,您可以检查以上几点,看看是否有误,并尝试调整相应的参数或硬件资源。
阅读全文
收起全文