2 回答

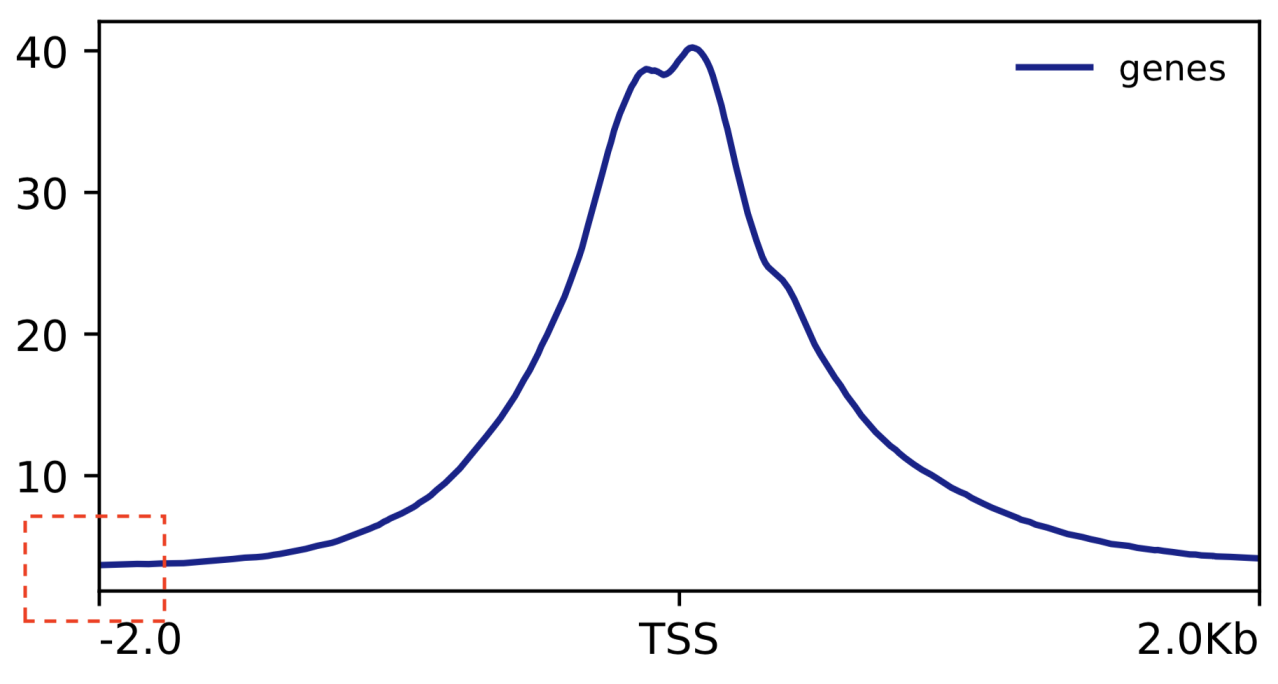
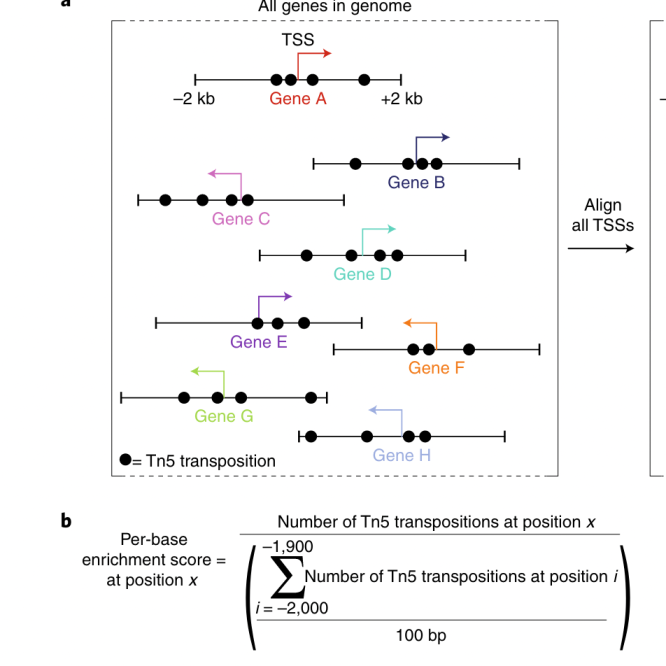
首先,TSS enrichment score是用来衡量某个基因的转录起始位点(TSS)在ATAC-seq数据中的富集程度的。计算TSS enrichment score时,通常会将基因的TSS定义为起点,然后计算在一定距离范围内(例如2kb)ATAC-seq信号的富集程度。
对于问题中提到的情况,纵坐标不是从1开始的,这可能是由于computeMatrix计算过程中使用的是默认参数,将所有信号值都转换为正数。因此,得到的图中信号值的最小值可能不是1。
为了确保TSS enrichment score的计算正确,可以通过以下步骤进行:
通过以上步骤,可以确保得到的TSS enrichment score是正确的,同时也可以保证纵坐标从1开始。