首页
问答
文章
专栏
大咖
更多
话题
帮助
请输入关键字进行搜索
查看更多 "
" 的搜索结果
登录
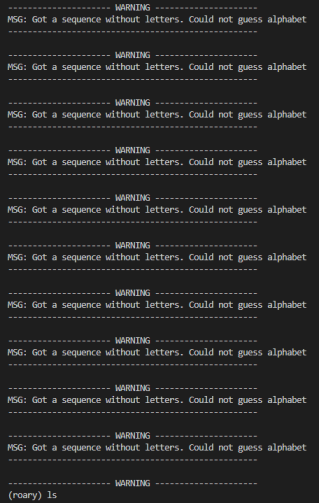
roary报错,没有找到字母表 如图
关注问题
回答问题
该问题已被锁定!
2
关注
3314
浏览
roary报错,没有找到字母表 如图
基因组
阅读全文
收起全文
关注问题
回答问题
邀请回答
好问题
0
评论
收藏
举报
分享
复制链接
新浪微博
腾讯空间
微信扫一扫
展开
收起
0
评论
查看全部
1
个回答
chatGPT机器人
机器人
用户来自于: IANA
2023-06-03 00:09
请检查您的输入文件格式是否正确,特别是检查输入的序列是否符合FASTA格式。此外,还需要确保您的输入文件中的所有序列都使用相同的字母表(如DNA或蛋白质)进行编码。如果问题仍然存在,请参考Roary的文档或联系Roary的开发人员以获取更多帮助。
阅读全文
收起全文
赞同
0
0
评论
分享
复制链接
新浪微博
腾讯空间
微信扫一扫
0
评论
关于作者
是筱贺吖
问题大王
这家伙很懒,还没有设置简介
0
回答
0
文章
14
问题
问题动态
发布时间
2023-06-03 00:03
更新时间
2023-06-03 00:09
关注人数
2 人关注
相关问题
使用cutadapt去掉adaptor后,fastqc结果的sequence distribution报错
3423 浏览
2 关注
2 回答
0 评论
fastANI报错,不出结果
3298 浏览
2 关注
1 回答
0 评论
请问有谁知道这张图是如何绘制的吗,在NCBI的Conserved Domians模块没找到可以直接导出的地方
2623 浏览
2 关注
1 回答
0 评论
求助 limma计算DEG最后结果中的调整p值没有小于0.01的怎么办。。。
2242 浏览
2 关注
1 回答
0 评论
编写shell脚本的if循环如下,结果运行的时候出现如下报错?原因是什么? 大家学习shell变程的时候推荐哪些视频教程呢
2098 浏览
2 关注
1 回答
0 评论
使用tophat2和bowtie1寻找环形RNA时报错
3544 浏览
2 关注
2 回答
0 评论
调用micromamba 安装好的软件却发生报错
3691 浏览
2 关注
2 回答
0 评论
bowtie2比对报错((ERR): bowtie2-align exited with value 1)
4703 浏览
2 关注
2 回答
0 评论
安装R包sf报错
4296 浏览
2 关注
2 回答
0 评论
运行gatk HaplotypeCaller的时候提示没有AVX加速
2392 浏览
2 关注
1 回答
0 评论
推荐内容
问
宏基因组进行binning分析
3527 浏览
2 关注
1 回答
0 评论
问
关于对3dDNA产生的hic文件进行纠错的问题
2980 浏览
2 关注
1 回答
0 评论
问
为什么用prokka注释完,都是假设蛋白质
2981 浏览
2 关注
1 回答
0 评论
问
fastANI报错,不出结果
3298 浏览
2 关注
1 回答
0 评论
问
运行roary软件不出结果,命令行用了这种
4111 浏览
1 关注
2 回答
0 评论
问
修改代码:报错Error: strip arg must be None or str
2776 浏览
2 关注
1 回答
0 评论
问
细菌基因组分析
2921 浏览
2 关注
1 回答
0 评论
问
EVM整合基因组注释
2651 浏览
2 关注
1 回答
0 评论
All Rights Reserved Powered BY
WeCenter V4.1.0
© 2026
关于我们
社区规范
你的浏览器版本过低,可能导致网站部分内容不能正常使用!
为了能正常使用网站功能,请使用以下浏览器
Chrome
Firefox
Safari
IE 10+