learn_graph(cds)绘制plot_cells(cds, color_cells_by = "cell.type")正常,
但在在order_cell(cds)命令选择自定义节点后,运行plot_cells(cds,color_cells_by="pseudotime)后报错:
Error in value[[3L]](cond) :
No pseudotime for UMAP calculated. Please run order_cells with reduction_method = UMAP before attempting to color by pseudotime.
服务器和本地R上都尝试了,但报错结果相同。希望路过的朋友指点一下。
以下是完整代码,在最后一步绘图报错,
文中cds.rds数据链接: https://pan.baidu.com/s/12JTZYiWOFPc95MIE3WSwNg?pwd=e5wb
################################# Information ##################################
# Single Cell Analysis (processed matrix)
#
#
# Log: 2023-9-25 start writing;
# 2023-9- accomplish version 1.0;
# 2023-9-25 last update.
#
# Data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132257
# Ref: https://www.nature.com/articles/s41588-020-0636-z
# https://cole-trapnell-lab.github.io/monocle3/
################################################################################
#### Lib Packages ####
library(Seurat)
library(monocle3)
library(ggplot2)
library(dplyr)
#### set work path ####
rm(list = ls())
setwd("~/Code/RCode/SingleCell/RefDemo/Demo2/GSE132257/")
#################################### START #####################################
library(monocle3)
library(Seurat)
#### 1) Input data #############################################################
filepath1 <- "seurat.rds"
seurat <- readRDS(filepath1)
data <- GetAssayData(seurat, assay = "RNA", slot = "counts")
Cell_metadata <- seurat@meta.data
Gene_annotation <- data.frame(gene_short_name = rownames(data), row.names = row.names(data))
cds <- new_cell_data_set(data,
cell_metadata = Cell_metadata,
gene_metadata = Gene_annotation)
#### 2) Process data ###########################################################
# normalize, calculate dimension
cds <- preprocess_cds(cds, method = "PCA", num_dim = 100,
scaling = TRUE)
dev.new()
plot_pc_variance_explained(cds)
dev.off()
# Reduce the dimensions using UMAP
cds <- reduce_dimension(cds, reduction_method = "UMAP",
umap.fast_sgd = T, cores = 5)
dev.new()
plot_cells(cds, color_cells_by = "seurat_clusters")
dev.off()
dev.new()
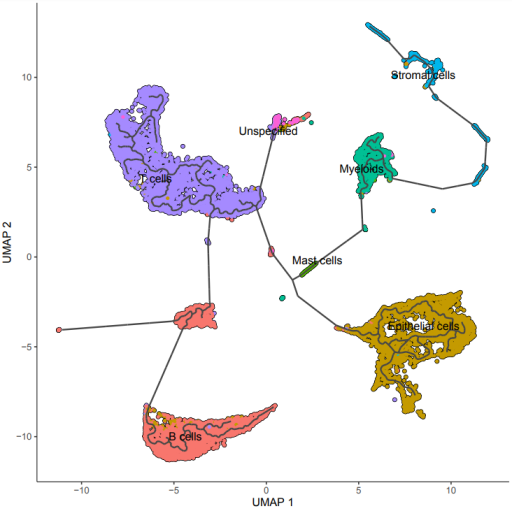
plot_cells(cds, reduction_method = "UMAP", color_cells_by = "cell.type")
plot_cells(cds, reduction_method = "UMAP", color_cells_by = "orig.ident")
dev.off()
# Remove batch effects
cds <- align_cds(cds, alignment_group = "orig.ident", num_dim = 100)
cds <- reduce_dimension(cds, reduction_method = "UMAP",
umap.fast_sgd = T, cores = 5)
# cluster
cds <- cluster_cells(cds, resolution = 1e-5)
dev.new()
plot_cells(cds, reduction_method = "UMAP", color_cells_by = "cell.type")
plot_cells(cds, reduction_method = "UMAP", color_cells_by = "orig.ident")
dev.off()
#### 3) Trajectory in pseudotime ###############################################
cds <- learn_graph(cds)
dev.new()
plot_cells(cds,
color_cells_by = "cell.type",
label_groups_by_cluster = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE,
group_label_size = 4,
cell_size = 1.0)
dev.off()
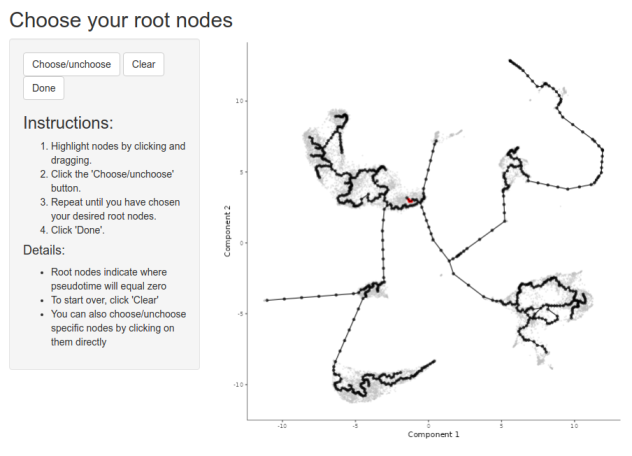
order_cells(cds, reduction_method = "UMAP")
# 绘图报错!报错!报错!
dev.new()
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5,
group_label_size=4,
cell_size=1.0)
dev.off()
#### Save cds ##################################################################
saveRDS(cds, file="cds.rds")
##################################### END ######################################
