如何将转录本某个m6a修饰的位置坐标,转换成基因组坐标。
有尝试biostars上的一些解决方法,也都转换成功,但是最终得出的基因组坐标信息与RMbase数据库中的修饰信息做overlap,得到的数量很少。数据库50w左右的修饰位点,鉴定出2w多的位点仅有1k多overlap。所以我认为是在转换坐标上出现了问题,但是又不知道具体问题在哪,下面是我的代码:
txdb <- makeTxDbFromGFF("gencode.v43.annotation.gtf",format = "gtf")
gr <- GRanges(seqnames = data$ENST, ranges = IRanges(start=data$start,
end=data$end, names = data$ENST), strand = "*")
gr
# isolate transcripts and genes from txdb
transcripts <- transcripts(txdb)
mcols(transcripts)$tx_name <- transcripts$tx_name
names(transcripts)<-mcols(transcripts)$tx_name
# use mapFromTranscripts in the GenomicFeatures package
map2genome <- mapFromTranscripts(gr, transcripts)
另外转换出的坐标与在基因组上看,对应的碱基都不是A或者T,有人知道是什么问题吗?
2 回答
关于转录组坐标转基因组坐标,我给你个神器。
这个是我之前的一位师兄写的,现在人已经在兰大独立当PI了。
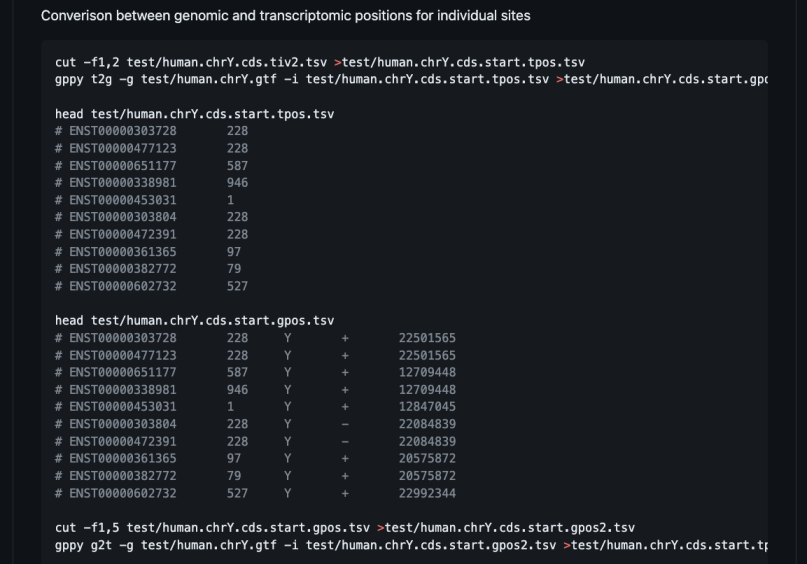
https://github.com/mt1022/gppy
主要功能:
python小工具,用于GTF文件信息提取,转bed12,基因组与转录本坐标互相转换。仅依赖python标准包。