请输入关键字进行搜索
查看更多 "" 的搜索结果
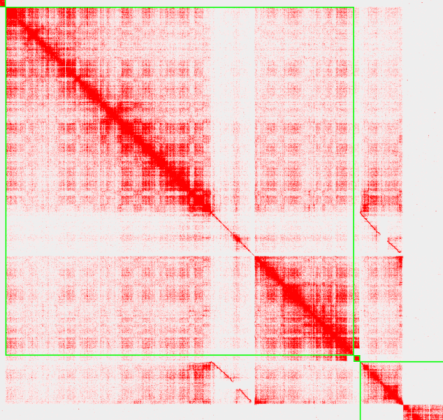
对3dDNA产生的hic文件,使用juicerbox进行纠错的时候,发现出现图中的错误类型。
查看全部 1 个回答
这家伙很懒,还没有设置简介
你的浏览器版本过低,可能导致网站部分内容不能正常使用!
为了能正常使用网站功能,请使用以下浏览器