请教大佬们,我有十几个小样本的scRNA,每个只有小几千个细胞,使用harmony默认参数整合后批次效应还是很明显,想请教一下如果针对很多样本的整合,harmony有什么参数可以调整的吗?或者其他可以接Seurat的包更适合多样本的吗?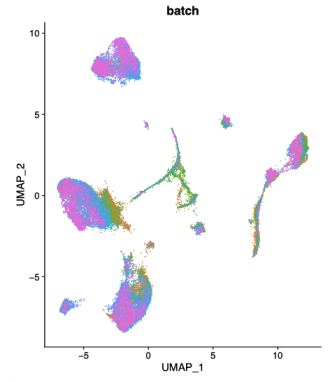
1 回答

关于多个scRNA样本整合的问题,我建议您可以尝试以下方法:
综上所述,整合多个scRNA样本是一个复杂的问题,需要综合考虑多个因素,包括样本质量、数据预处理、整合方法等。希望您能够根据具体情况选择合适的方法,获得更好的整合效果。